When Killian Sheriff PhD ’26 sat down to simulate a nickel-cobalt-chromium alloy, he wasn’t just running code—he was unlocking a faster, more accurate way to predict how metals behave at the atomic level. At MIT’s Department of Materials Science and Engineering, Sheriff and his team have cracked a persistent problem: modeling chemically disordered metal alloys, the very materials that power jet engines, nuclear reactors, and high-performance electronics. For decades, scientists have struggled to simulate these complex materials because their atoms are arranged in unpredictable, ever-shifting patterns. Traditional methods required exhaustive computational brute force—sometimes over 100,000 hours per material—and still failed to capture real-world behavior accurately. Now, with a breakthrough in machine learning, the team led by Rodrigo Freitas, MIT’s TDK Career Development Professor, has built a smarter path forward.
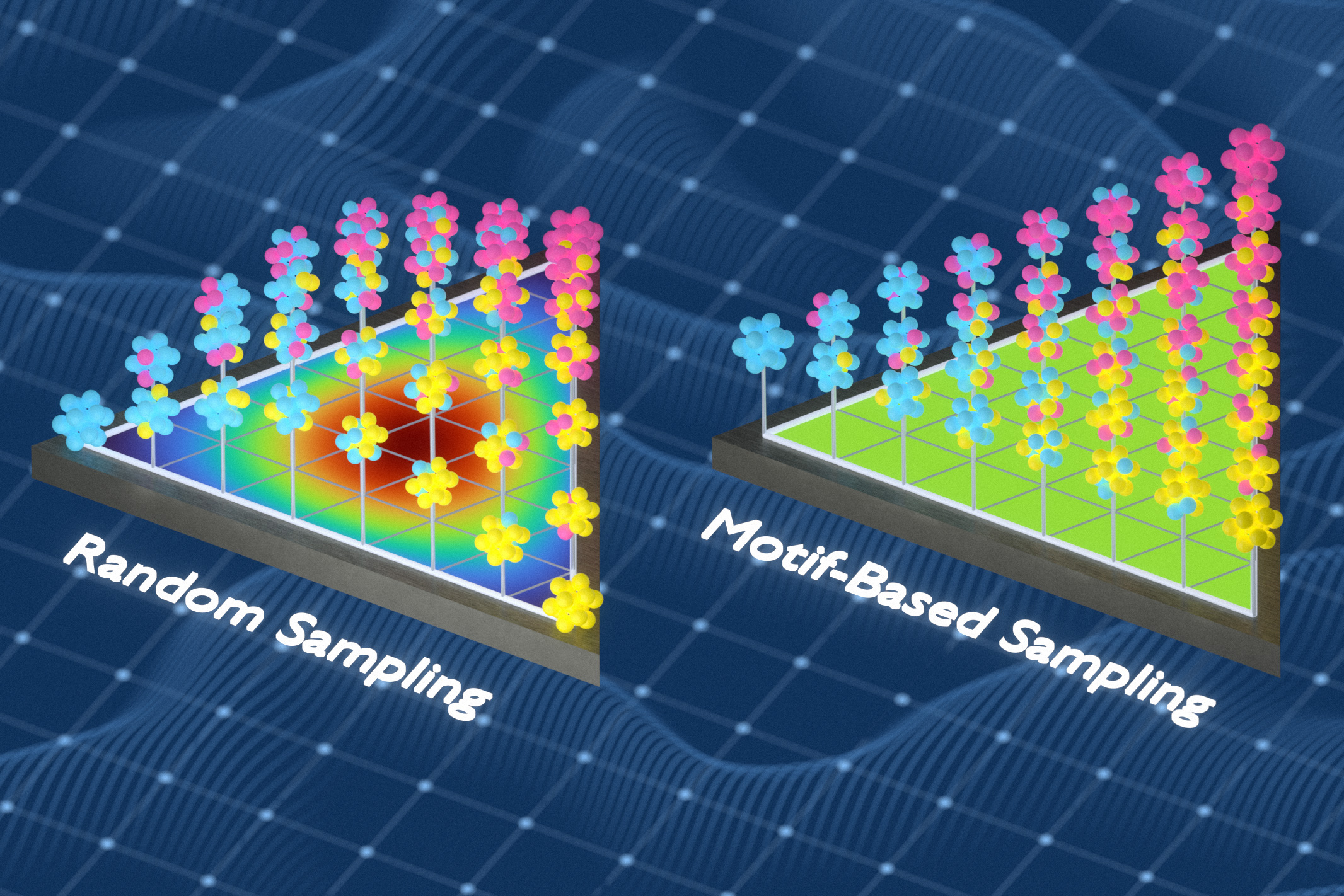
The innovation lies not just in the model, but in the data used to train it. Most machine-learning models for materials rely on simulations of atomic interactions, but their accuracy depends on how well the training data represents the chaotic diversity of atomic environments in disordered alloys. The MIT team used information theory—a mathematical framework for measuring uncertainty and diversity—to design training datasets that maximize the variety of local atomic configurations. By systematically swapping atoms in simulated structures, they eliminated redundant examples and ensured each data point taught the model something new. “We kept optimizing the training set so it captured as many different local environments as possible,” Freitas explains. The result? Models that learn more from less, achieving higher fidelity with fewer simulations.
Published in Science Advances, the study demonstrated that models trained on these optimized datasets outperformed those using random sampling or other standard methods in predicting key material properties like elastic stiffness and thermal expansion across a range of conditions. The team tested their approach on complex alloys including Ni-Co-Cr and high-entropy alloys, where traditional simulations often fall short. Crucially, the method reduces reliance on costly and time-consuming physical experiments—especially valuable when developing materials for extreme environments like fusion reactors or deep-space missions.
Beyond metals, the implications stretch into semiconductors, sustainable steels, and next-generation batteries. “This is not specific to any one application—you could use this approach to create new sustainable steels, new materials for aerospace, and more,” Freitas says. With collaborators including MIT PhD students Daniel Xiao and Yifan Cao, and University of Sheffield’s Lewis R. Owen, the team is already expanding the framework to other material classes. As industries race to innovate under tighter environmental and economic constraints, this method offers a powerful shortcut: simulating the real world not just faster, but better.