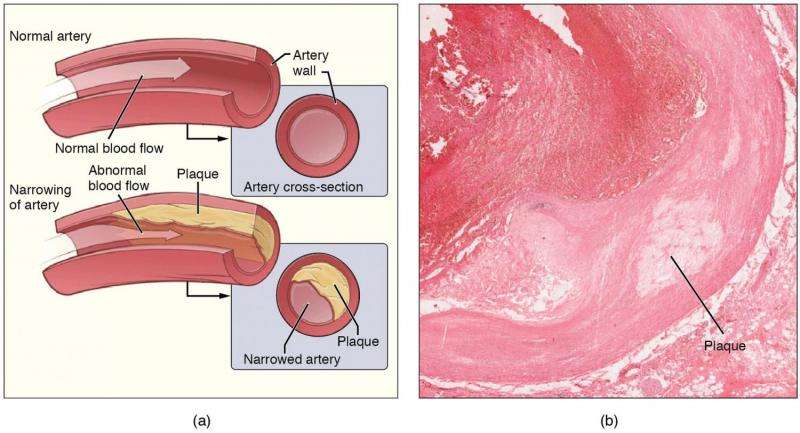
In the branching points where blood vessels split, cholesterol quietly accumulates in the arterial walls—a process that over years hardens into the dangerous plaques of atherosclerosis. Now researchers at LMU University Hospital in Munich have revealed a surprising twist: the immune system's response to these deposits is not simply destructive, but paradoxically protective in critical ways, opening a path toward therapies that could one day prevent heart attacks and strokes.
Atherosclerosis develops as the heart and brain's oxygen supply faces constant threat. When fatty deposits narrow or block vessels, the results can be catastrophic—angina pectoris, sudden blockages, or worse. The immune system's scavenger cells, called macrophages, accumulate in vessel walls over decades, engulfing fats and cholesterol. When these lipid-laden cells die, they leave behind cellular debris and cholesterol crystals that destabilize plaques and promote dangerous blood clots.
But a team led by Professor Andreas Schober and Dr. Maliheh Nazari Jahantigh discovered that a second type of macrophage—those that remain lipid-free—fundamentally shapes the disease's progression. Using four-dimensional microscopic imaging of plaques in mice, the researchers found these cells perform a delicate balancing act. They clear away the DNA and cellular debris left by dead cells, which slows cholesterol crystal formation and helps limit plaque growth. Simultaneously, however, these same macrophages attack the thin protective lining inside blood vessels, the endothelium. Inflammation, remarkably, acts as both a damaging force and a limiting one.
At the center of this biological balance sits a small RNA molecule: miR-147. Produced mainly in lipid-free macrophages, this microRNA enables the cells to remove dead cell debris while simultaneously restraining damage to the endothelium. The research team showed that when miR-147 is absent, plaque formation increases markedly, along with dangerous accumulations of DNA from dead cells and cholesterol crystals. The mechanism reveals why: miR-147 suppresses production of a protein called Galectin-3. When Galectin-3 is released unchecked, it damages endothelial cells and disrupts the macrophages' energy supply, leaving them unable to clear away debris efficiently—a failure that accelerates plaque buildup.
"The inflammatory response in atherosclerosis is complex and includes both harmful effects and mechanisms that limit plaque growth," said Dr. Nan Li, the study's first author. "This is precisely where a therapeutic opportunity lies: a treatment based on miR-147 could selectively influence inflammatory processes in atherosclerotic plaques and, in the long term, reduce the risk of heart attack and stroke."
The findings, published in Circulation in 2026, suggest a remarkably precise intervention point. Rather than broadly dampening inflammation—which could be dangerous—future therapies could harness miR-147 to amplify the immune system's own protective mechanisms within plaques. By enhancing the lipid-free macrophages' ability to clear debris while limiting endothelial damage, such treatments could tip the balance decisively in favor of plaque stability. For millions living with atherosclerosis, this shift from blunt anti-inflammatory approaches to targeted molecular manipulation represents a genuine path forward.