In the fight against heart disease, researchers at UT Southwestern Medical Center have spotted a previously hidden switch—a protein called HELZ2 that sits deep inside liver cells, controlling how many cholesterol-carrying particles slip into the bloodstream. The discovery, published in the American Heart Association journal Circulation, reveals a fundamentally new way to regulate cholesterol before it even becomes a problem.
For decades, doctors have relied on statins to fight harmful cholesterol after it's already circulating through the body. But this finding points to an earlier intervention point: controlling the genetic instructions that tell liver cells to manufacture apoB proteins in the first place. Those apoB proteins form lipoproteins—the particles that transport cholesterol and fats throughout the body and eventually stick to artery walls, building the plaque that leads to heart attacks and strokes.
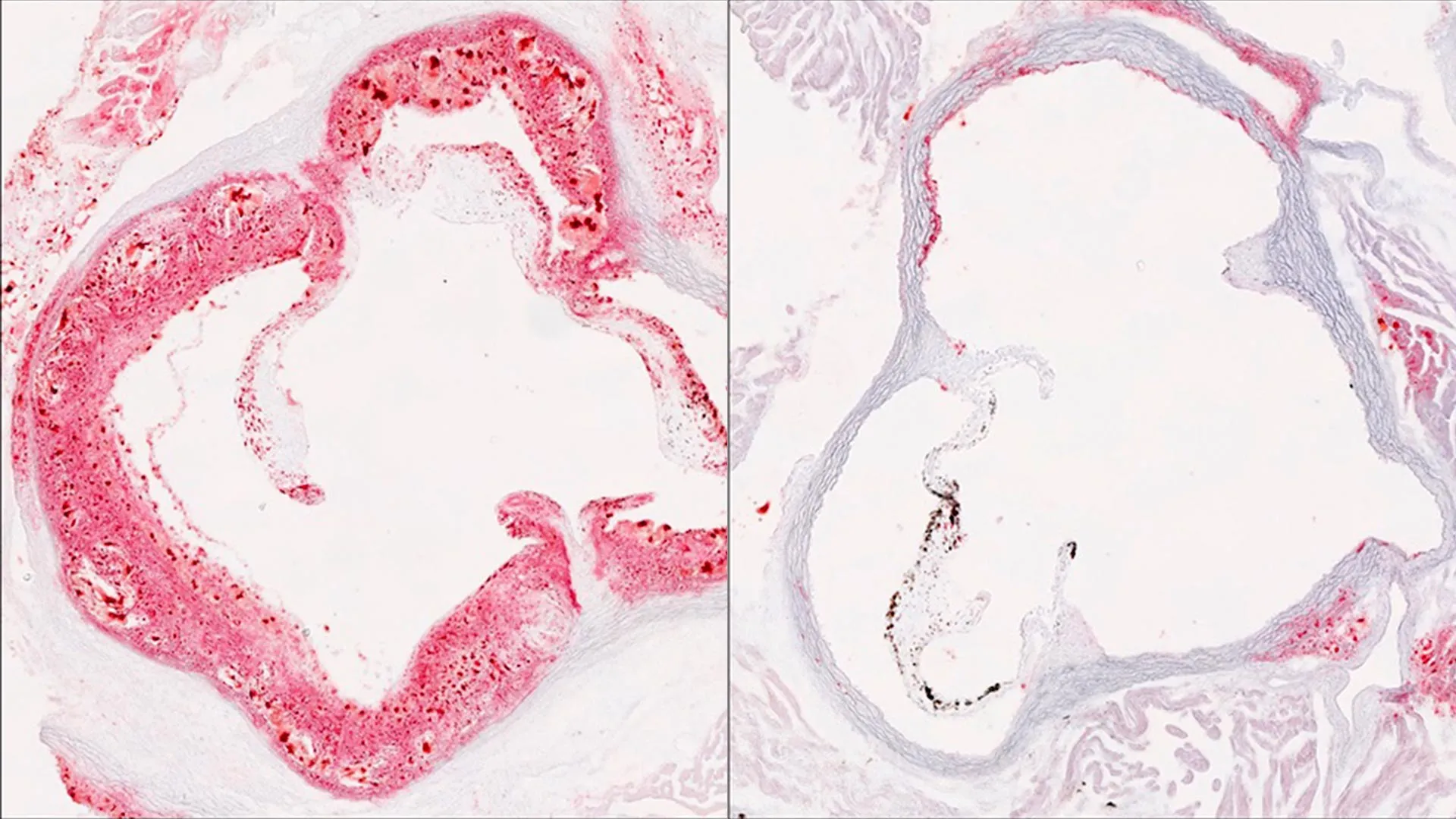
Zhao Zhang, Ph.D., an Assistant Professor in UT Southwestern's Center for the Genetics of Host Defense and of Internal Medicine, led the team in uncovering HELZ2's surprising role. "These particles are a major driver of plaque buildup in the arteries," Zhang explained. "What we found is that HELZ2 acts as a powerful control point for how many cholesterol-carrying particles ultimately enter the bloodstream."
The research hinged on a creative approach. Using a large-scale genetic screening system developed by Nobel Prize winner Bruce Beutler, M.D., the team studied unusual fat buildup in the livers of mice and identified a gain-of-function mutation that increased HELZ2 activity. When HELZ2 activity rose, something unexpected happened: it shortened the lifespan of apoB messenger RNA—the genetic instruction inside liver cells that tells them to make apoB proteins. When those instructions break down faster, fewer apoB proteins get made, and fewer cholesterol-carrying particles enter the blood.
Yiao Jiang, Ph.D., a postdoctoral researcher in the Zhang Lab, highlighted what made this discovery surprising: "Most previous research focused on what happens to apoB after it's already made. What surprised us is that HELZ2 acts much earlier, by controlling how long the apoB 'message' survives before the protein is even produced."
The mice carrying the HELZ2 mutation produced fewer lipoproteins overall, including LDL cholesterol and triglycerides, in their bloodstream. They also showed greater protection against atherosclerosis, the artery-clogging disease that causes heart attacks and strokes. But the research also revealed an important trade-off: when blood cholesterol dropped in these animals, more fat accumulated in their livers. Mice without the mutation showed the opposite pattern—more cholesterol in the blood but less fat in the liver.
This delicate balance is precisely what makes HELZ2 interesting as a therapeutic target. "We can think of HELZ2 as a kind of dial between the liver and the bloodstream," Zhang said. "Turning it up lowers cholesterol in the blood but increases liver fat. Turning it down does the reverse. That balance makes HELZ2 especially interesting as a potential therapeutic target."
The insight could reshape treatment strategies. Rather than targeting cholesterol after it's produced—the mechanism behind statins—carefully adjusting HELZ2 activity might offer a new way to reduce dangerous cholesterol levels while potentially opening new approaches to treating fatty liver disease. "The idea that we can control apoB at the RNA level represents a major shift in how we think about cholesterol regulation," Zhang said. "It gives us a new molecular lever—and potentially a new set of tools—for tackling these conditions."